
By Bethann Hromatka, Ph.D.
Myeloproliferative neoplasms (MPN) have a complicated name but it’s an apt descriptor for the biology underlying this group of rare blood disorders.
Blood Cells and Bone Marrow
MPNs result from excessive production of “myeloid” blood cells that originate in the bone marrow.
They come in different forms, including essential thrombocythemia (ET), polycythemia vera (PV), myelofibrosis (MF), chronic myeloid leukemia (CML), and systemic mastocytosis. Most forms of MPN are cancerous, and all types are rare, with each affecting fewer than 100,000 Americans.
In 1951, a physician named William Dameshek proposed the idea of myeloproliferative disorders. Still, it wasn’t until 2005 – when four articles were published back-to-back on the V617F mutation in the JAK2 gene – that researchers began to uncover clues about the genetic basis of these diseases.
The V617F Mutation
Discovered in blood cells of people with MPN, the V617F mutation is defined as a somatic or acquired mutation, which means that it arises during a person’s life as opposed to being a change in DNA that someone is born with. (See sidebar for more about the JAK2 V617F mutation and its clinical use.)
JAK2 in the Clinic
The JAK2 protein is a tyrosine kinase and plays important roles in the cell by directing the activity and movement of other proteins. Although more research is needed to clearly define the role of the JAK2 V617F mutation in myeloproliferative disease, there is strong evidence that cells bearing this mutation are less prone to dying and are very good at making multiple copies of themselves, a hallmark of cancer. The World Health Organization (WHO) has recommended that the JAK2 V617F mutation be used as a diagnostic marker of disease and numerous companies are developing drugs to inhibit the activity of JAK2, though it is too early to tell if they will be successful.
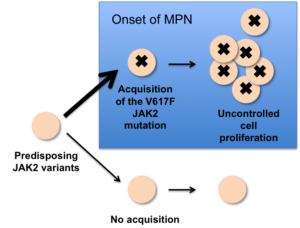
This figure shows that people who inherit predisposing JAK2 variants have higher odds of acquiring the JAK2 V617F mutation, which is thought to push the cell to make multiple copies of itself. But it’s not the only factor leading to MPN. Although most people with the polycythemia vera (PV) form of MPN have the V617F mutation, many people with other types of MPN do not.
In addition to the V617F mutation, several studies, performed mainly in people with European ancestry, have shown that inherited genetic factors may also increase the risk for MPN. A cluster of SNPs in the JAK2 gene, the 46/1 or GGCC JAK2 haplotype, has been associated with MPN and seems to be a predisposing factor for acquiring the V617F somatic mutation. People with a G at (equivalent to in the study) had nearly four times higher odds of developing V617F-positive MPN than those without the G version. 23andMe recently replicated this association. However, we see a smaller effect – in our database, people with a G have about two times the odds of developing V617F-positive MPN compared to people without the G version.
The Japanese Cohort
A more recent study with a Japanese cohort suggests that the association between relatively common variants in the JAK2 gene and MPN also applies to individuals with Asian ancestry.
Junko Ohyashiki and colleagues from Tokyo Medical University report that people with an A at in the JAK2 gene (equivalent to those reported in the study and highly correlated with ) have about four times higher odds of developing V617F-positive MPN compared to individuals without the A version.
When they looked more closely, they found even stronger evidence for the association and a larger effect in patients with the polycythemia vera (PV) form of MPN, which is consistent with the fact that over 95% of people with PV have the V617F mutation. In contrast, only about half of people with essential thrombocythemia (ET) or myelofibrosis (MF) have V617F. The SNP did not appear to be linked to V617F-negative MPN.
Looking Ahead
Although these findings are still preliminary since they were collected from relatively small studies (Ohyashiki’s findings came from just 95 people with MPN), they are still exciting progress in research on these rare disorders. That relatively common genetic variation might predispose individuals to acquire other mutations during their lifetime is an intriguing concept requiring more research.
Editor’s note: This post has been edited from the original to reflect changes to our product.


